固定电势法计算 OER
模拟恒电势的催化反应过程,需要配合溶剂模型使用。 更详细的视频教程可以从官网获取。 整个计算流程有以下几步:
note
方式一:
step1: 催化剂模型构建+固定电势法结构优化(晶格优化+原子弛豫) → 总能;
step2: 中间体模型构建+固定电势法结构优化(原子弛豫)→ 总能 → 零点能计算;
step3: 气体分子模型构建+结构优化(原子弛豫)→ 总能 → 零点能计算;
step4: 查表得到熵值(例如NIST-JANAF Thermochemical Tables);
step5: 计算吉布斯自由能。
方式二:
step1: 催化剂模型构建+结构优化(晶格优化+原子弛豫) → 固定电势法自洽计算 → 总能;
step2: 中间体模型构建+结构优化(原子弛豫)→ 固定电势法自洽计算 → 总能 + 零点能计算;
step3: 气体分子模型构建+结构优化(原子弛豫)→ 自洽计算 → 总能 + 零点能计算;
step4: 查表得到熵值;
step5: 计算吉布斯自由能。
以 C3N 掺杂 Co 原子吸附 OH-为例子(方式二-step2)。构建吸附模型结构并进行后续计算,点击组件→系统组件→ 拖拽结构优化和自洽计算组件到工作流区域 → 添加从结构导入到结构优化再到自洽计算的连线。其中结构导入组件中导入的是 C3N 掺杂 Co 原子吸附 OH-的结构。
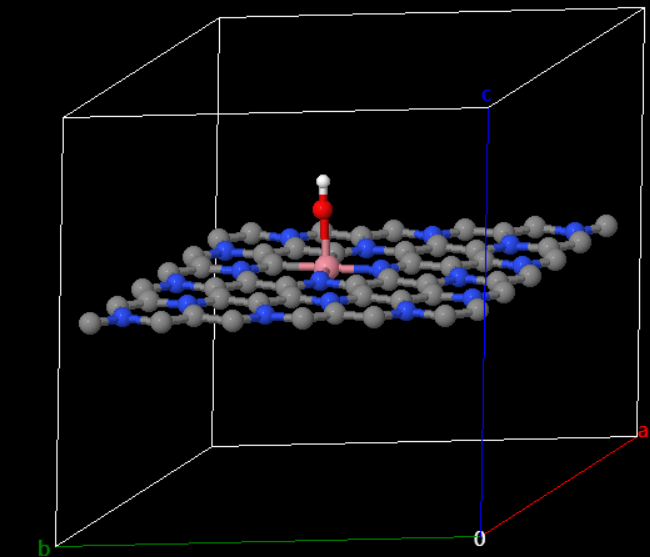
note
自洽计算时考虑溶剂效应,并进行一些设置。在此基础上,还需要开启离子浓度设置和固定电势计算。
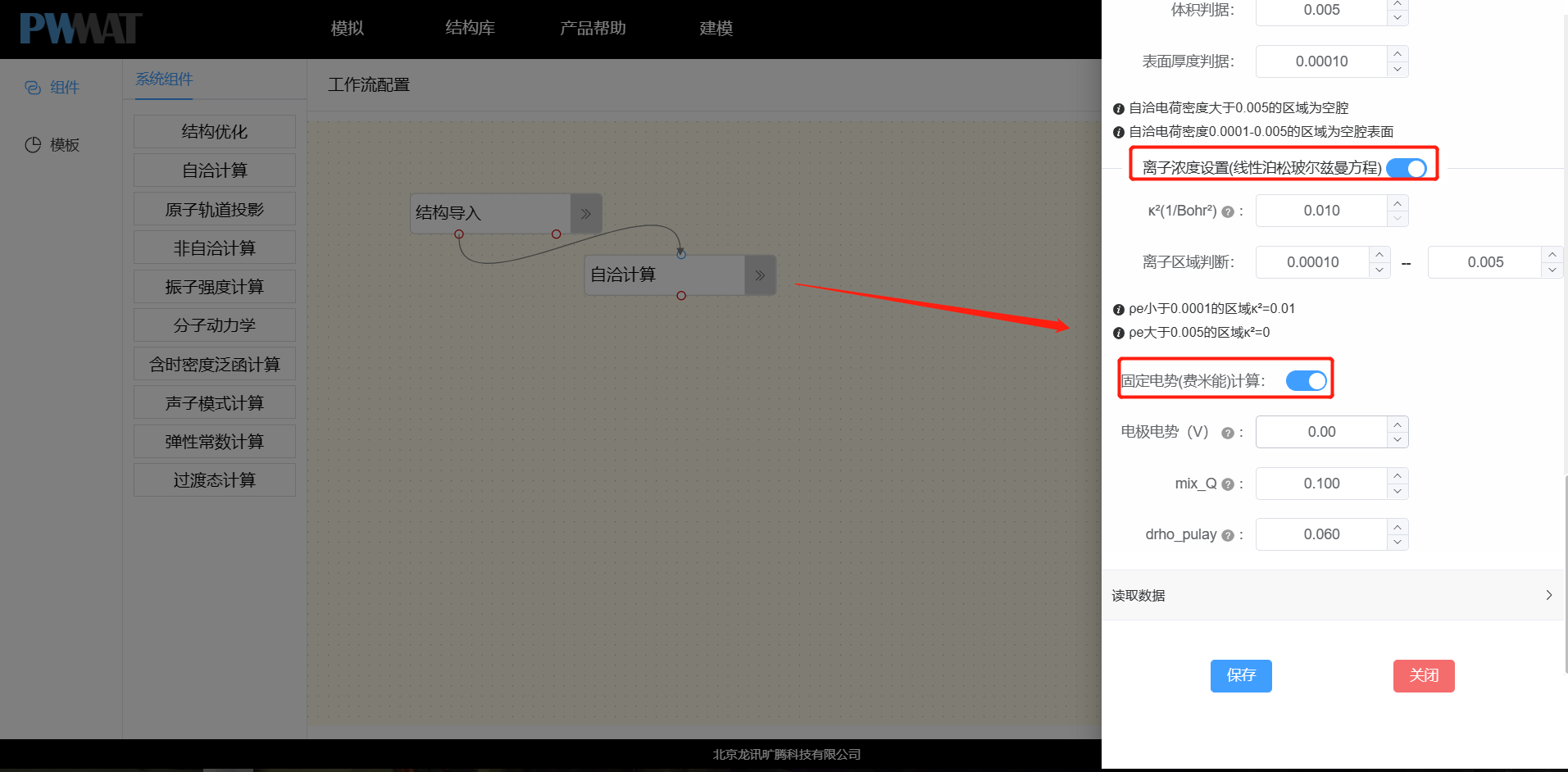
关于固定电势计算需要注意:
- 设置目标电极电势,这里是以标准氢电极为参考系,即相对标准氢电极的值;
- 收敛情况不好时可以适当增加
mix_Q和drho_pulay值;
计算时可以拖拽多个自洽计算组件到工作流区,为不同的自洽计算组件分别设置不同的电极电势。后续的其他计算的操作,与溶剂模型计算中相同。
最后从各个组件中提取数据根据公式计算吉布斯自由能。