Bulk Cu system
The following section provides an example of how to use the PWMLFF Deep Potential Model for training and LAMMPS simulation using a Cu system.
The overall program workflow is roughly divided into:
1. Generate Dataset
Using Cu data obtained from PWmat AIMD simulations as an example, the data files are MOVEMENT300 and MOVEMENT1500, each containing 100 structures with 72 Cu atoms per structure.
Example etot.input file:
8 1
JOB = MD
MD_DETAIL = 2 100 1 300 300
XCFUNCTIONAL = PBE
ECUT = 60
ECUT2 = 240
MP_N123 = 2 2 3 0 0 0 3
IN.ATOM = atom.config
IN.PSP1 = Cu.SG15.PBE.UPF
ENERGY_DECOMP = T
OUT.STRESS = F
- Optional
ENERGY_DECOMP: Whether to decompose the total DFT energy into atomic energies. Results are output in theMOVEMENTfile. Set toTif atomic energies are needed for use or training. - Optional
OUT.STRESS: Whether to output stress information. Set toTif trainingVirialis required. - For other parameter meanings, refer to the PWmat manual.
2. Training the Force Field
2.1 Processing Dataset
Create a new *.json file (e.g., extract.json) in the working directory. This file is used to call pwdata for processing molecular dynamics trajectory files and extracting labels.
Example:
{
"valid_shuffle": true,
"train_valid_ratio": 0.8,
"raw_files": ["./MOVEMENT300", "./MOVEMENT1500"],
"format": "pwmat/movement"
}
Where:
valid_shuffle: Whether to randomly shuffle all data. For example, if the molecular dynamics step size is 10 and there are 10 images, withvalid_shuffleset totrue, the 10 images will be shuffled randomly and then split into training and validation sets according totrain_valid_ratio. Ifvalid_shuffleisfalse, the data will be split sequentially according totrain_valid_ratio. Default isTrue.train_valid_ratio: Ratio of training to validation sets.raw_files: Path to the raw data.format: Format of the raw data used for generating the training set. Supported formats includemovement,outcar,cp2k/md.
Run the command pwdata extract.json to generate a PWdata folder in the current directory, containing train and valid subfolders with training and validation data.
Then, modify the force field training input control file *.json (e.g., dp_cu.json) to specify the datasets_path where the label files are located. (See below)
2.2 Input File
In the current directory, the force field training input control file includes several parameters.
Example input file (Other parameters for input files):
{
"raw_files": ["/Cu/PWdata/Cu72", "/Cu/PWdata/Cu72_1"],
"model_type": "DP",
"atom_type": [29]
}
datasets_path: Path to the label files. Multiple paths can be specified, each containing training and validation subdirectories. Adjust as needed.model_type: Type of model used for training. For other model types and parameter configurations, refer to Parameter Details.atom_type: Atomic type, where the atomic number of Cu is 29.
2.3 Running
The following slurm example script is suitable for Mcloud. Ensure that the necessary environment and modules are loaded before submitting the job.
#!/bin/sh
#SBATCH --partition=3090
#SBATCH --job-name=mlff
#SBATCH --nodes=1
#SBATCH --ntasks-per-node=1
#SBATCH --gres=gpu:1
#SBATCH --gpus-per-task=1
#Mcloud installed environment loaded
# Recommended here
source /share/app/PWMLFF/PWMLFF2024.5/env.sh
# Alternatively, the following method can be used for step-by-step loading
# source /share/app/anaconda3/etc/profile.d/conda.sh
# module load conda/3-2020.07
# conda deactivate
# conda activate PWMLFF
# module load pwmlff/2024.5
PWMLFF train dp_cu.json > log
For interactive running:
$ srun -p 3090 --pty /bin/bash
#Mcloud installed environment loaded
$ source /share/app/PWMLFF/PWMLFF2024.5/env.sh
$ PWMLFF train dp_cu.json
In most cases, you can use the raw_files parameter to directly call pwdata for data processing and training:
In this case, you can skip running pwdata extract.json separately and run PWMLFF train dp_cu.json directly. For example:
{
"raw_files": ["./MOVEMENT300", "./MOVEMENT1500"],
"format": "pwmat/movement",
"valid_shuffle": true,
"train_valid_ratio": 0.8,
"model_type": "DP",
"atom_type": [29]
}
During training, you can check the training status by looking at the logs in the directory where the model files are stored (model_record).
This directory contains the following files:
dp_model.ckptis the model file, which can be used to continue training or extract the force field. It corresponds to the most recent training model.epoch_train.datandepoch_valid.datlog files contain training and validation errors for each epoch.
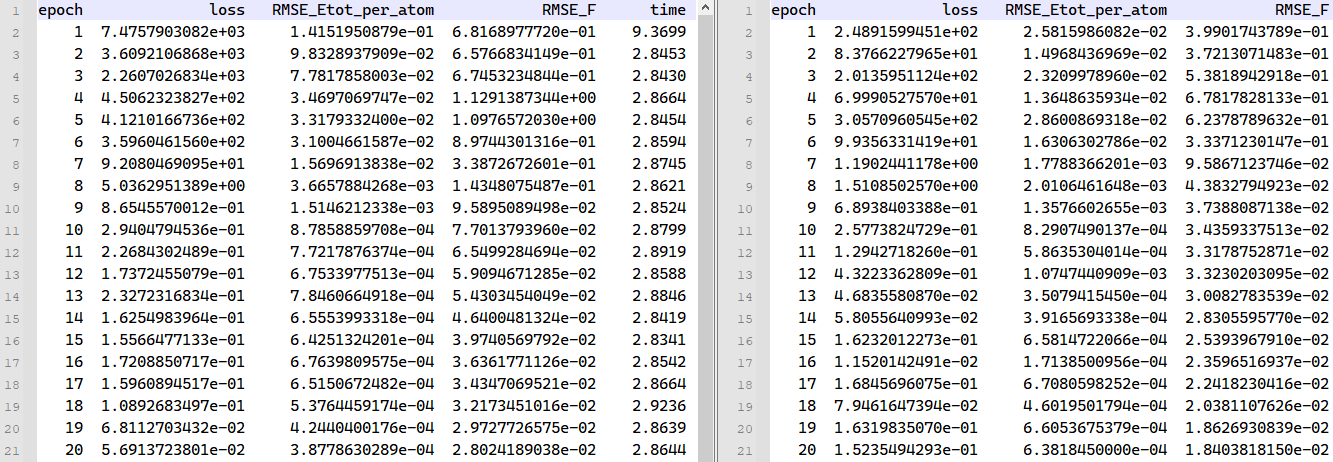
losscorresponds to the total training error.RMSE_Etot_per_atomcorresponds to the energy error in training, with a suggested target of around ~.RMSE_Fcorresponds to the force error in training, with a suggested target of around ~.
2.4 Extracting Force Field
It is recommended to use the Libtorch version of the force field model. After training, manually execute the PWMLFF script dp_model.ckpt command to generate the jit_dp_cpu.pt or jit_dp_gpu.pt file. This file is used for LAMMPS simulation.
If your device includes a GPU environment, executing PWMLFF script will generate the jit_dp_gpu.pt file; otherwise, it will generate the jit_dp_cpu.pt file.
Note: jit_dp_gpu.pt can only run LAMMPS in a GPU environment; jit_dp_cpu.pt can only run LAMMPS in a CPU environment.
After training is complete, the default behavior is to generate a forcefield folder in the current directory, containing *.ff force field files. This force field file should be used with the specific version, compiled, and used according to the previous manual.
3. LAMMPS Simulation
Use the pt force field file generated after training for LAMMPS simulation.
To use the force field file generated by PWMLFF, an example LAMMPS input file is as follows:
pair_style pwmlff 1 ../model_record/jit_dp_gpu.pt
pair_coeff * * 29
Where:
pair_style pwmlff 1indicates using the PWMLFF generated force field file, with1indicating reading one force field file.../model_record/jit_dp_gpu.ptis the PWMLFF generated force field file; adjust the path as needed.pair_coeff * * 29specifies the atomic number for Cu.
Here is an example LAMMPS input file (NVT ensemble):
units metal
boundary p p p
atom_style atomic
processors * * *
neighbor 2.0 bin
neigh_modify every 10 delay 0 check no
read_data lmp.init
pair_style pwmlff 1 ../model_record/jit_dp_gpu.pt
pair_coeff * * 29
velocity all create 1500 206952 dist gaussian
timestep 0.001
fix 1 all nvt temp 1500 1500 0.1
thermo_style custom step pe ke etotal temp vol press
thermo 1
dump 1 all custom 1 traj.xyz id type x y z vx vy vz fx fy fz
run 1000 #1ps
-
When running LAMMPS with GPU, use the executable
lmp_mpi_gpu; when running with CPU, uselmp_mpi. -
If there are multiple force field files (e.g., during active learning), such as 4 files, you can modify it to:
pair_style pwmlff 4 1.pt 2.pt 3.pt 4.pt
pair_coeff * * 29
4. Other Input File Parameters Explanation
{
"recover_train": false,
"raw_files": ["0_300_MOVEMENT", "1_500_MOVEMENT"],
"format": "pwmat/movement",
"valid_shuffle": true,
"train_valid_ratio": 0.8,
"model_load_file": "./model_record/dp_model.ckpt",
"model_type": "DP",
"atom_type": [29],
"max_neigh_num": 100,
"seed": 1234,
"model": {
"descriptor": {
"Rmax": 6.0,
"Rmin": 0.5,
"M2": 16,
"network_size": [25, 25, 25]
},
"fitting_net": {
"network_size": [50, 50, 50, 1]
}
},
"optimizer": {
"optimizer": "LKF",
"block_size": 5120,
"kalman_lambda": 0.98,
"kalman_nue": 0.9987,
"nselect": 24,
"groupsize": 6,
"batch_size": 4,
"epochs": 20,
"start_epoch": 1,
"print_freq": 10,
"train_energy": true,
"train_force": true,
"train_ei": false,
"train_virial": false,
"train_egroup": false,
"pre_fac_force": 2.0,
"pre_fac_etot": 1.0,
"pre_fac_ei": 1.0,
"pre_fac_virial": 1.0,
"pre_fac_egroup": 0.1
}
}
recover_train: Whether to continue training from where it was last interrupted or completed. If set totrue, the program will read from the defaultmodel_load_pathandmodel_nameto resume training from the last checkpoint. See Parameter Details.raw_files: Path and names of the molecular dynamics trajectory files. Multiple files can be specified. Modify according to the actual situation.train_valid_ratio: Ratio of training set to validation set.0.8means 80% of the data is used for training and 20% for validation.model_load_file: Path to the model file. If specified, the program will read from this path and continue training/testing from the specified model file. See Parameter Details.model_type: Type of model currently used for training. For training and parameter configurations of other model types, refer to Parameter Details.atom_type: Atomic type, where the atomic number for Cu is 29.max_neigh_num: Maximum number of neighboring atoms.seed: Random number seed.model: Model parameters. For specific parameter configurations, refer to Parameter Details.optimizer: Optimizer parameters, recommended areLKFandADAM. Generally, for large systems and networks, using theLKFoptimizer can speed up training. For other optimizers and more parameter configurations, refer to Parameter Details.batch_size: Size of data used per batch for training. For example, 1, 2, 5, 10.n_epoch: Number of training iterations. Adjust according to the total number of dynamics trajectory images. For fewer images, the number of epochs can be increased, e.g., to 50.