Features (or descriptors) are quantities that describe the local atomic environment of an atom. They are required preserve the translational, rotational, and permutational symmetries. Features are usually used as the input of various regressors(linear model, NN, .etc), which output atomic energies and forces.
Features are differentiable functions of the spatial coordinates, so that force can be calculated as
Fi=−dRidEtot=−j,α∑∂Gj,α∂Ej∂Ri∂Gj,α
where :j is the index of neighbor atom within the cutoff radius, and :α the index of feature.
Additionally, features are required to be rotionally, translationally, and permutaionally invariant.
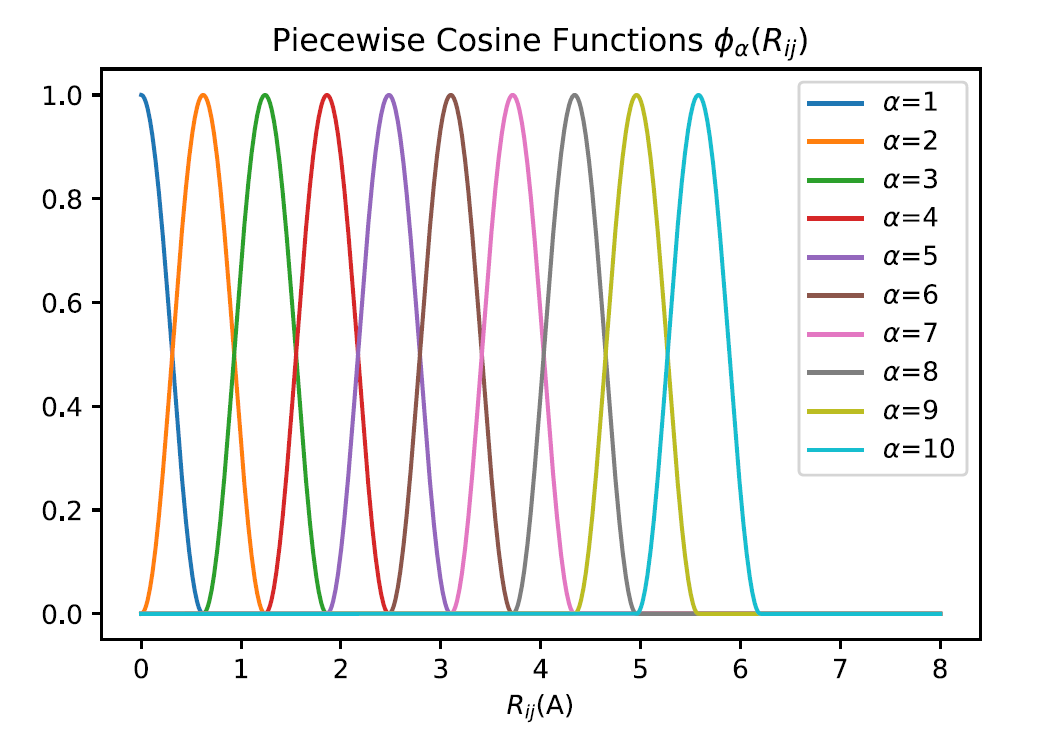
2-b and 3-b features with piecewise cosine functions (feature 1 & 2)
Given a center atom, the piecewise cosine functions are used as the basis to describe its local environment. The praph below gives you an idea of how they look like.
We now define the pieceswise cosine functions, in both 2-body and 3-body feaures. Given the inner and outer cut Rinner and Router, the degree of the basis M, the width of piecewise function h, and the interatomic distance between the center atom i and the neighbor jRij, one defines the basis function as
The expression of 2-b feature with center atom i is thus
Gα,i=m∑ϕα(Rij)
and 3-b feature
Gαβγ,i=j,k∑ϕα(Rij)ϕβ(Rik)ϕγ(Rjk)
where ∑m and ∑m,n sum over all atoms within cutoff Router of atom i
In practice, these two features are usually used in pair.
Reference:
Huang, Y., Kang, J., Goddard, W. A. & Wang, L.-W. Density functional theory based neural network force fields from energy decompositions. Phys. Rev. B 99, 064103 (2019)
These two are the features first used in Behler-Parrinello Neural Network. Given the cutoff radius Rc, and the interatomic distance Rij with center atom i, define cutoff function fc
and η, ζ, and λ=±1 are parameters defined by user.
In practice, these two features are usually used in pair.
Reference:
J. Behler and M. Parrinello, Generalized Neural-Network Representation of High Dimensional Potential-Energy Surfaces. Phys. Rev. Lett. 98, 146401 (2007)
In MTP, the local environment of the center atom :math:i is characterized by
ni=(zi,zj,rij)
where zi is the atom type of the center atom, zj atom type of the neighbor j, and rij the relative coordinates of neighbors. Next, energy contribution of each atom is expanded as
Ei(ni)=α∑cαBα(ni)
where Bα are the basis functions of choice and cα the parameters to be fitted.
We now introduce moment tensors Mμν to define the basis functions
Mμν(ni)=j∑fμ(∣rij∣,zi,zj)ν⨂rij
These moments contain both radial and angular parts. The radial parts can be expanded as
fμ(∣rij∣,zi,zj)=β∑cμ,zi,zj(β)Q(β)(∣rij∣)
where Q(β)(∣rij∣) are the radial basis funtions. Specifically,
where ϕ(β) are polynomials (e.g. Chebyshev polynomials) defined on the interval [Rmin,Rcut]
The angular part ⨂νrij, which means taking tensor product of rijν times, contains the angular information of the neighborhood ni. ν determines the rank of moment tensor. With ν=0 one gets a constant scalar, ν=1 a vector (rank-1 tensor), ν=2 a matrix (rank-2 tensor), .etc.
Define further the level of moments as
lev(Mμν)=2+4μ+ν
This is an empirical formula.
Reference
I.S. Novikov, etal, The MLIP package: moment tensor potential with MPI and active learning. Mach. Learn.: Sci. Technol, 2, 025002 (2021)
In SNAP, it does not use Gaussian progression. So it does not calculate the distance and kernel between two atomic local environment maps. It first defines a atomic local environment, then use sphoerical harmonic (or 4D sphere, with rotation matrix) to expand the atomic local environment. It then uses the bispectrum, so it becomes rotational invariant. In a way it is like the MTP, but it uses a special way to contract out the orientational index to make it roatational invariant. It is often used with linear regression.
Consider the local environment of neighboring atoms around the central atom i at position r as a sum of δ functions in a three-dimensional space:
ρ(r)=δ(r)+rki<RC∑fC(rki)ωkδ(r−rki)
where rki is the position of the kth neighbor of atom i, ωk is the weight of the kth neighbor, and the radial function fC(rki) ensures that the contribution of each neighbor will smoothly vary to zero near the cutoff radius RC:
fC(r)=0.5[cos(RCπr)+1]
The angular part of this local environment can be expanded in the familiar basis of spherial harmonic functions defined for l=0,1,2,... and m=−l,−l+1,...,l−1,l. The radial distribution is usually represented by a set of radial basis functions. However, here, the radial information r is mapped into another angle information in a 4D hyper spherical function Umm′j(θ0,θ,ϕ), where all the points (neighboring atoms) fall into a 3D spherical surface (in a 4D space) and the orientational (angual) information is given by the there angles:
r≡xyz→ϕ=arctan(y/x)θ=arccos(z/r)θ0=43πr/rc
As a result, the above local environment function can be expanded by these 4D hyper spherical harmonics Umm′j(θ0,θ,ϕ) with an expansion coefficient umm′j:
Here, k is the neighboring atom index, and θ0(k),θ(k),ϕ(k) are the three angles of the position vector rki of the kth neighbor of atom i. Note, umm′j is the orientational dependent due to its index m,m′. They can be contracted out (using three umm′j) with the following contraction formula:
Here, Cj1m1j2m2jmCj1m1′j2m2′jm is the Clebsch-Gordan coefficient, and the final scalar features are F(j1,j2,j). We can produce different features by setting diffrent j1,j2,j. Note, in these features, there is no radial function index, instead we have three angual momentum indexex. This is because we have converted the radial distance information into the third angle information in a 3D sphere.
Here, RC2 is a smooth cutoff parameter that allows the components in ri to smoothly go to zero at the boundary of the local region definded by RC. This smooth function is a bit more complex than the previous smooth function using a RC2. S(rji) reduce the weight of the atom further away from the center atom i. We then define radial functions gM(s) as the Chebyshev polynomial CM in the deep potential-Chebyshev features:
gM(s)=CM(2RminS−1).
Here, Rmin is an input for minimum r.
To construct the DeepMD feature, we next calculate the four components vector as:
TM(k)=rji<RC∑X^ji(k)S(rji)gM(S(rji))
Here, k=0,1,2,3 (four component vector). They are constructed from the usual x,y,z component, plus the S component as:
{xji,yji,zji}→{S(rji),x^ji,y^ji,z^ji}
where x^ji=rjiXji,y^ji=rjiYji,z^ji=rjiZji are the unit vectors of rji.
From these 4D vectors, one can contract the component index to yield a scalar feature as:
F(M1,M2)=k=0∑3TM1(k)TM2(k)
Here, M1 also encoded the number of atom types besides the Chebyshev. So, if the max Chebyshev order is M, the number of features is M∗ntype∗(M∗ntype+1)/2. We can produce different features by setting different M
This is much like in deep potential-Chebyshev, but we use Gaussian functions instead of using Chebyshev
polynomials, and the position and width parameters are specified by user.
Similar to deep potential-Chebyshev, the 4D vectors are constructed as:
M1 also encoded the number of atom types besides the Chebyshev. So, if the max Chebyshev order is M, the number of features is M∗ntype∗(M∗ntype+1)/2. We can produce different features by setting different M.