隐式溶剂效应下 OER 计算
关于溶剂化模型的视频教程可以从官网获取。 整个计算流程有以下几步:
step1: 催化剂模型构建+结构优化(晶格优化+原子弛豫) → 隐式溶剂模型自洽计算 → 总能;
step2: 中间体模型构建+结构优化(原子弛豫)→ 隐式溶剂模型自洽计算 → 总能 + 零点能计算;
step3: 气体分子模型构建+结构优化(原子弛豫)→ 自洽计算 → 总能 + 零点能计算;
step4: 查表得到熵值(例如NIST-JANAF Thermochemical Tables);
step5: 计算吉布斯自由能 。
以 C3N 掺杂 Co 原子吸附 OH-为例子。构建吸附模型结构并进行后续计算,点击组件→系统组件→ 拖拽结构优化和自洽计算组件到工作流区域 → 添加从结构导入到结构优化再到自洽计算的连线。其中结构导入组件中导入的是 C3N 掺杂 Co 原子吸附 OH-的结构。自洽计算时考虑溶剂效应,并进行一些设置。
关于溶剂效应需要注意的几个点 :
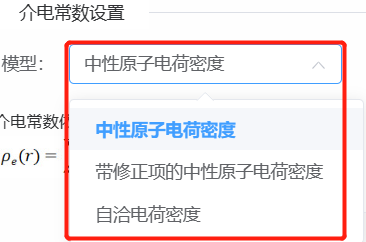
- 介电常数设置,介电常数通过电荷密度来区分溶质和溶剂,支持三种模型。
- 中性原子电荷密度表示使用赝势文件中的电荷密度(推荐使用);
- 带修正项的中性原子电荷密度表示对赝势中的电荷密度增加一个指数项;
- 自洽电荷密度表示使用自洽计算得到的电荷密度进行设置。
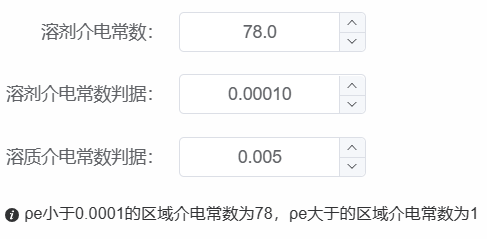
- �其中 f 列表示对应元素的权重,溶剂化效应强的元素,或电荷密度权重大时,设置大的值;溶剂介电常数默认为水的介电常数(78);溶剂介电常数判据和溶质介电常数判据为设置的电荷密度区间,电荷密度小于溶剂介电常数时为溶剂,电荷密度大于溶质介电常数时为溶质。计算完成后可视化溶剂化模型查看结果,检查判据设置是否合理,即能区分溶剂和溶质。

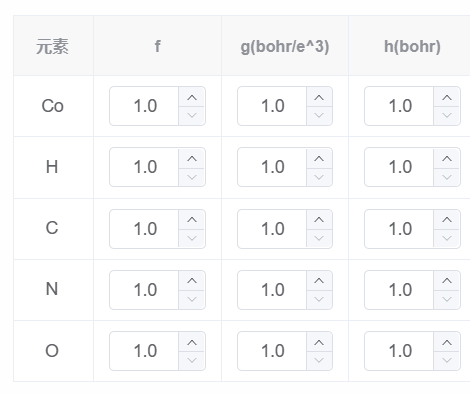
- 溶质腔设置,参数的设置影响溶剂化能量,表面张力和压强的参数设置依据文章或者实验值;体积判据使用了自洽计算的电荷密度区分溶质腔和溶剂。
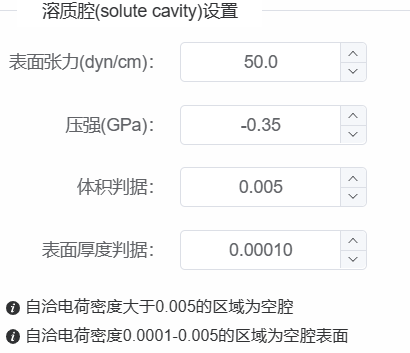
计算完成后点击自洽计算组件查看结果,可以查看溶剂效应模型。结果中可以区分出溶剂区、固液界面过渡区、溶质腔以及溶质区。如果溶质部分存在较大的孔洞,说明介电常数设置不合理,需要调整电荷密度区间或选择带修正项的中性原子电荷密度模型。
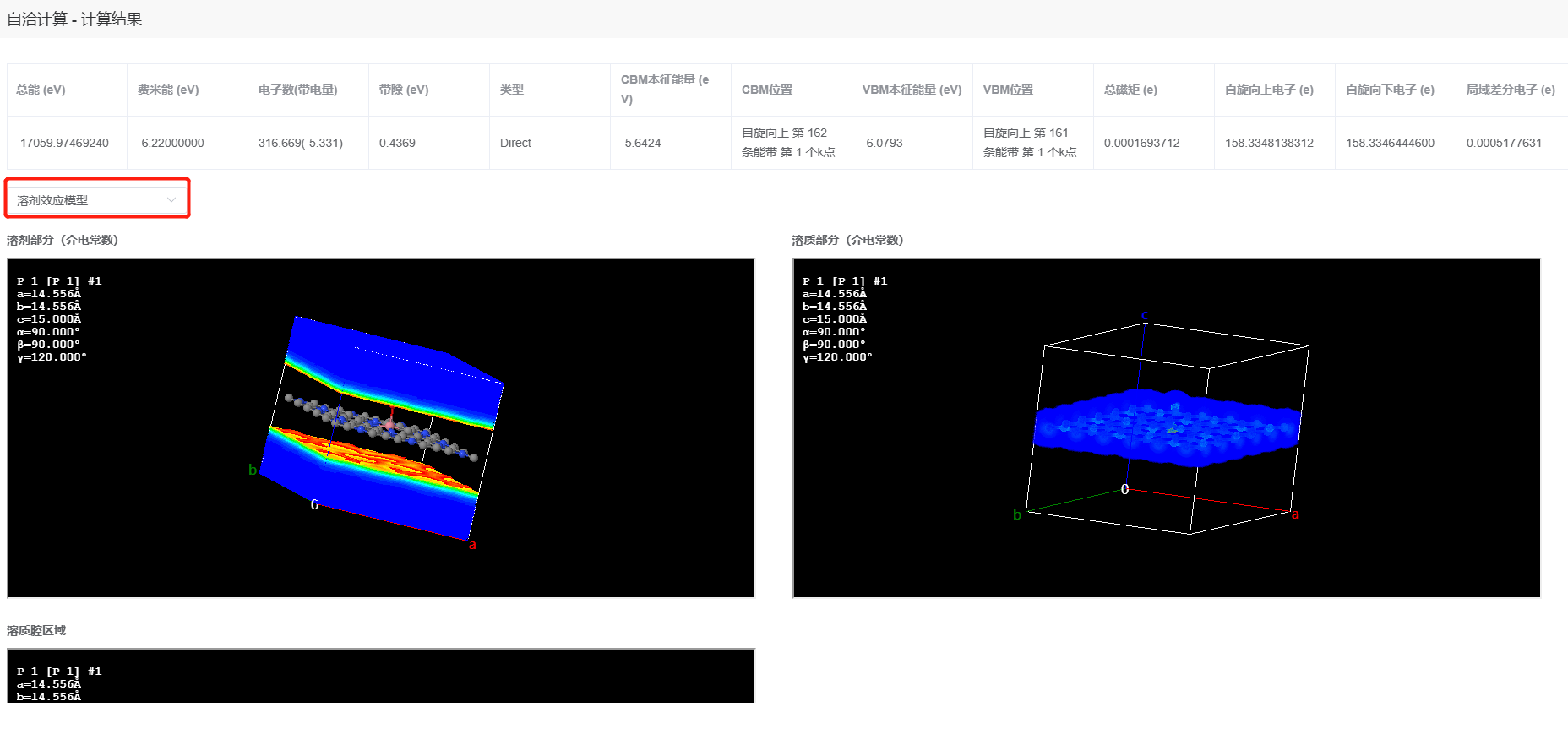
其中总能从自洽计算组件的结果中获取。

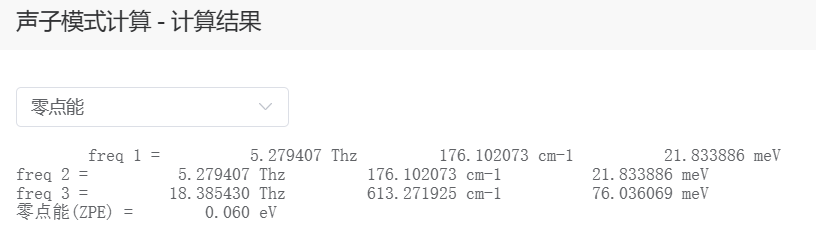
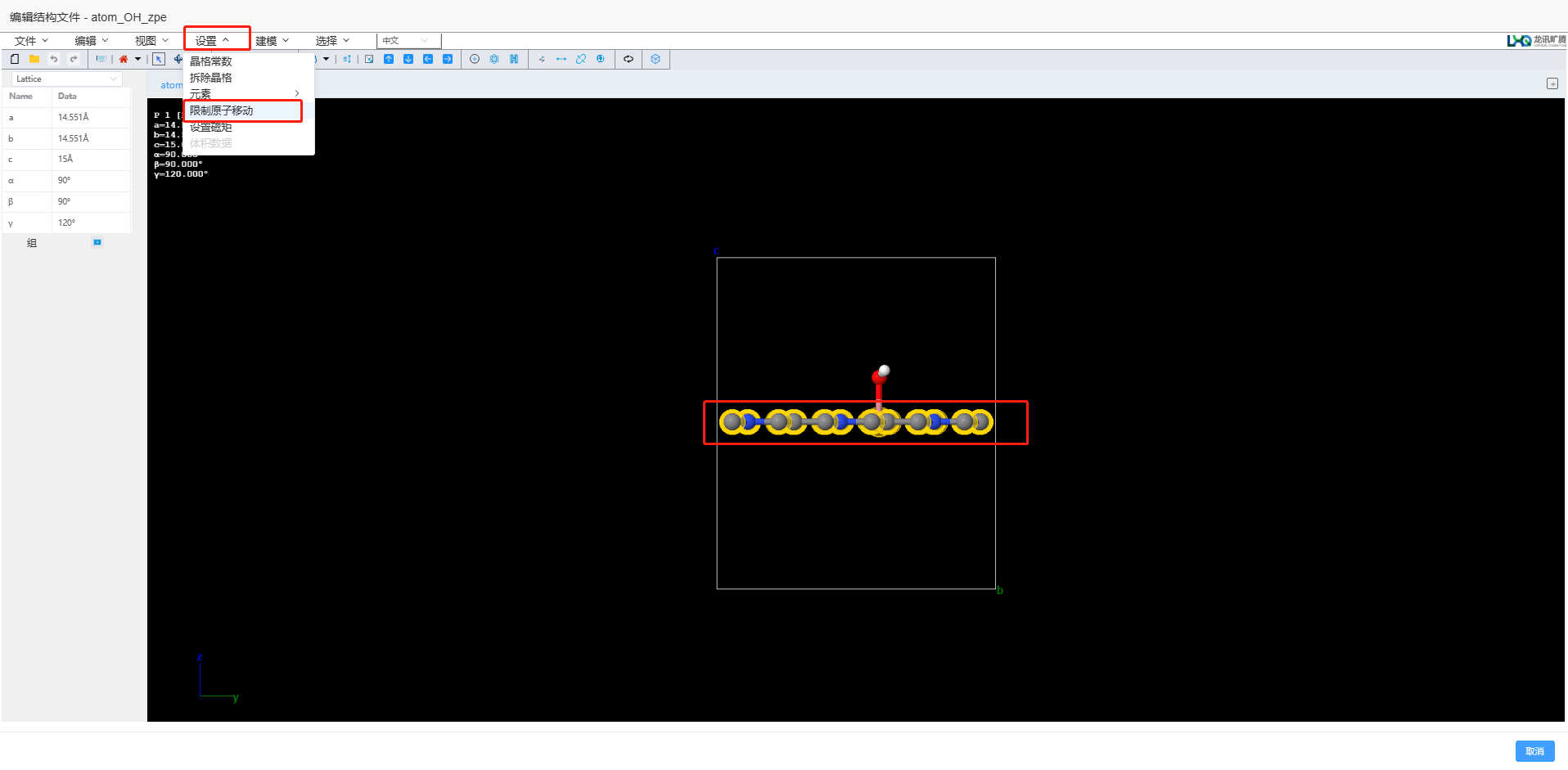
零点能的计算,对于吸附体系,只计算吸附分子的零点能,不计算基体,即固定催化剂部分(在 Q-Studio 中限制原子移动)。
回到结构库中,选择刚才用于自洽计算的结构 → 点击编辑,进入 Q-Studio,选择基体材料 → 点击设置 → 限制原子移动 → 导出结构。

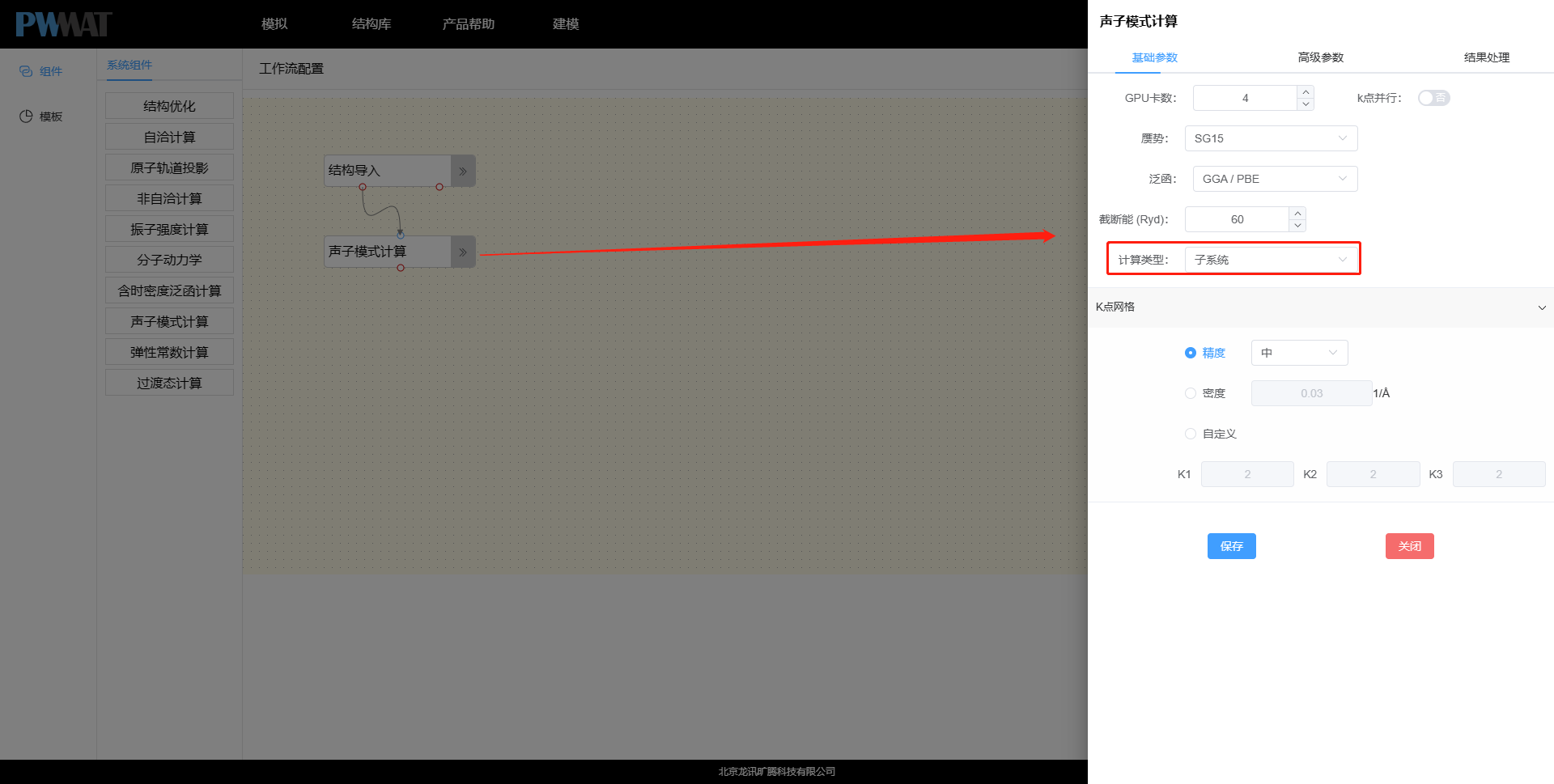
回到模拟中,并进入之前的项目 → 创建工作流 → 点击组件→系统组件→ 拖拽声子模式计算组件到工作流区域 → 添加从结构导入到到声子模式计算的连线。其中声子模式计算的参数设置中,计算类型选择子系统,即只对没有固定的吸附原子进行计算。
计算完成后点击声子模式计算组件查看结果,即可获取吸附分子的零点能。