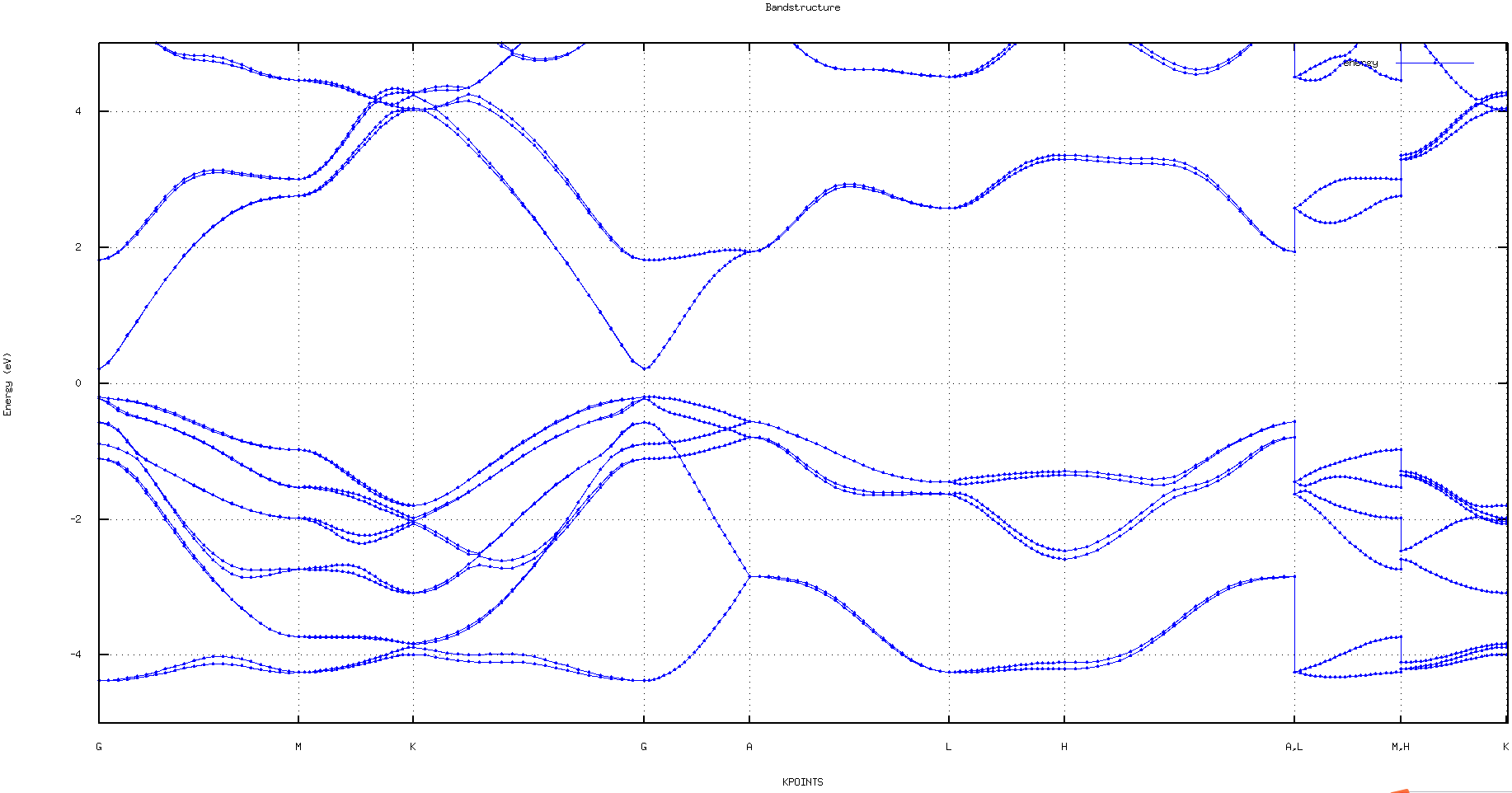
Spin-orbit coupling bandstructure calculation without magnetic moment
Spin-orbit coupling bandstructure calculation without magnetic moment for GaAs
There are two steps, the first step is SCF calculation, and the second is NONSCF calculation.
First Step: SCF calculation
Input files
atom.config
4
LATTICE
4.38647521 0.00000000 0.00000000
-2.19323810 3.79879878 0.00000000
0.00000000 0.00000000 7.15850229
POSITION
34 0.66666667 0.33333333 0.87584179 1 1 1
34 0.33333333 0.66666667 0.37584179 1 1 1
48 0.66666667 0.33333333 0.50004945 1 1 1
48 0.33333333 0.66666667 0.00004945 1 1 1
etot.input
1 4
JOB = SCF
IN.PSP1 = Cd.SG15.PBE.SOC.UPF
IN.PSP2 = Se.SG15.PBE.SOC.UPF
IN.ATOM = atom.config
CONVERGENCE = difficult
SPIN = 22
Ecut = 50
Ecut2 = 100
MP_N123 = 12 12 5 0 0 0
XCFUNCTIONAL = PBE
tip
- Spin: specifies spin polarization, 22:Spin-orbit coupling, but without magnetic moment.
- CONVERGENCE: control the convergence parameters of the SCF self-consistent iteration, possible values: easy or difficult.
Cd.SG15.PBE.SOC.UPF, Se.SG15.PBE.SOC.UPF
tip
Spin-orbit pseudopotential files need to be used.
Calculations
- You can submit PWmat tasks in different ways:
mpirun -np 4 PWmat | tee output
Note
Run the command directly
#!/bin/bash
#PBS -N SCF
#PBS -l nodes=1:ppn=4
#PBS -q batch
#PBS -l walltime=100:00:00
ulimit -s unlimited
cd $PBS_O_WORKDIR
mpirun -np 4 PWmat | tee output
Note
Submit the task with a pbs script
Second Step: NONSCF calculation
Input files
atom.config
4
LATTICE
4.38647521 0.00000000 0.00000000
-2.19323810 3.79879878 0.00000000
0.00000000 0.00000000 7.15850229
POSITION
34 0.66666667 0.33333333 0.87584179 1 1 1
34 0.33333333 0.66666667 0.37584179 1 1 1
48 0.66666667 0.33333333 0.50004945 1 1 1
48 0.33333333 0.66666667 0.00004945 1 1 1
etot.input
1 4
JOB = NONSCF
IN.PSP1 = Cd.SG15.PBE.SOC.UPF
IN.PSP2 = Se.SG15.PBE.SOC.UPF
IN.ATOM = atom.config
SPIN = 22
Ecut = 50
Ecut2 = 100
XCFUNCTIONAL = PBE
IN.VR = T
IN.KPT = T
Note
- Read IN.VR from previous SCF calculation. To copy OUT.VR and OUT.FERMI from the SCF calculation to your current working drectory and rename IN.VR.
- IN.KPT is the k-points file which PWmat will use for band structure calculation, one can use "split_kp.x" utility to get it. You should prepare an input file for "split_kp.x", which can be named "gen.kpt":
BAND # COMMENT line
20 # number of k-points between G and M
0.000 0.000 0.000 G # reciprocal coordinates; label 'G' for Gamma point
0.500 0.000 0.000 M
20
0.500 0.000 0.000 M
0.333 0.333 0.000 K
20
0.333 0.333 0.000 K
0.000 0.000 0.000 G
20
0.000 0.000 0.000 G
0.000 0.000 0.500 A
20
0.000 0.000 0.500 A
0.500 0.000 0.500 L
20
0.500 0.000 0.500 L
0.333 0.333 0.500 H
20
0.333 0.333 0.500 H
0.000 0.000 0.500 A
20
0.500 0.000 0.500 L
0.500 0.000 0.000 M
20
0.333 0.333 0.500 H
0.333 0.333 0.000 K
split_kp.x gen.kpt
After running "split_kp.x gen.kpt", it will generate "IN.KPT" and "HIGH_SYMMETRY_POINT" (high-symmetry points information) files.
Cd.SG15.PBE.SOC.UPF, Se.SG15.PBE.SOC.UPF
tip
Spin-orbit pseudopotential files need to be used.
Calculations
- You can submit PWmat tasks in different ways:
mpirun -np 4 PWmat | tee output
Note
Run the command directly
#!/bin/bash
#PBS -N SCF
#PBS -l nodes=1:ppn=4
#PBS -q batch
#PBS -l walltime=100:00:00
ulimit -s unlimited
cd $PBS_O_WORKDIR
mpirun -np 4 PWmat | tee output
Note
Submit the task with a pbs script
- After NONSCF calculation, you can run "plot_band_structure.x" to obtain band structure in your current working directory. Then it will generate the following files: bandstructure.eps, bandstructure.png, bandstructure.pdf and bandstructure_1.txt (the data file of band structure), which can be used to plot band by origin or gnuplot.
plot_band_structure.x